LAMMPS
LAMMPS 是大规模经典分子动力学代码,代表大规模原子/分子大规模并行模拟器。LAMMPS 具有用于软材料(生物分子、聚合物)、固态材料(金属、半导体)和粗粒度或细观系统的潜力。它可用于对原子进行建模,或者更一般地说,它可用作原子、中观或连续介质尺度上的平行粒子模拟器。
一. 模板提交
Step 1. 在应用中心搜索lammps软件,申请后请联系客服同意;
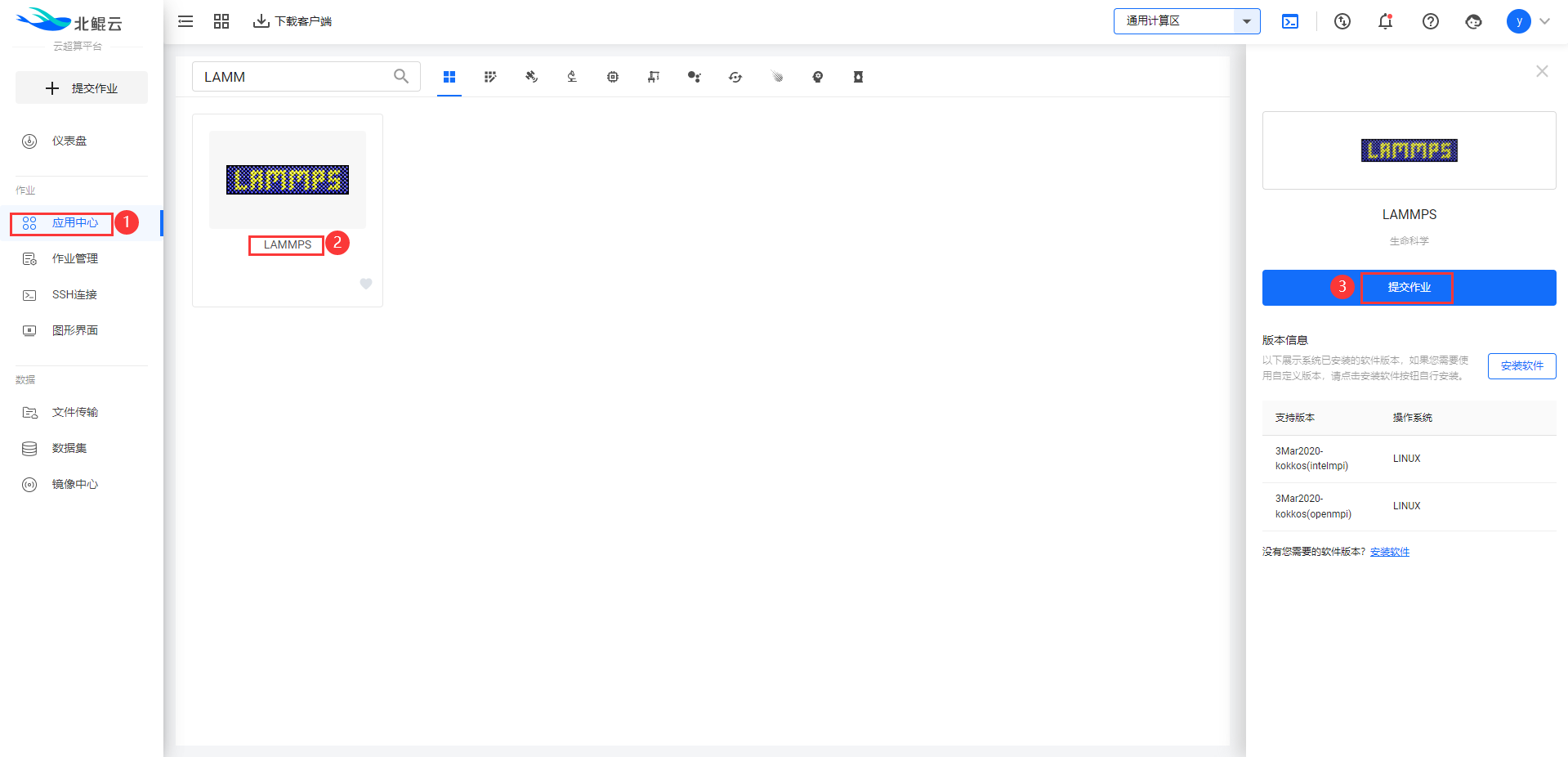
Step 2. 选择可视化模板;

Step 3. 点击输入文件列表上传文件,必须至少包含.in文件;
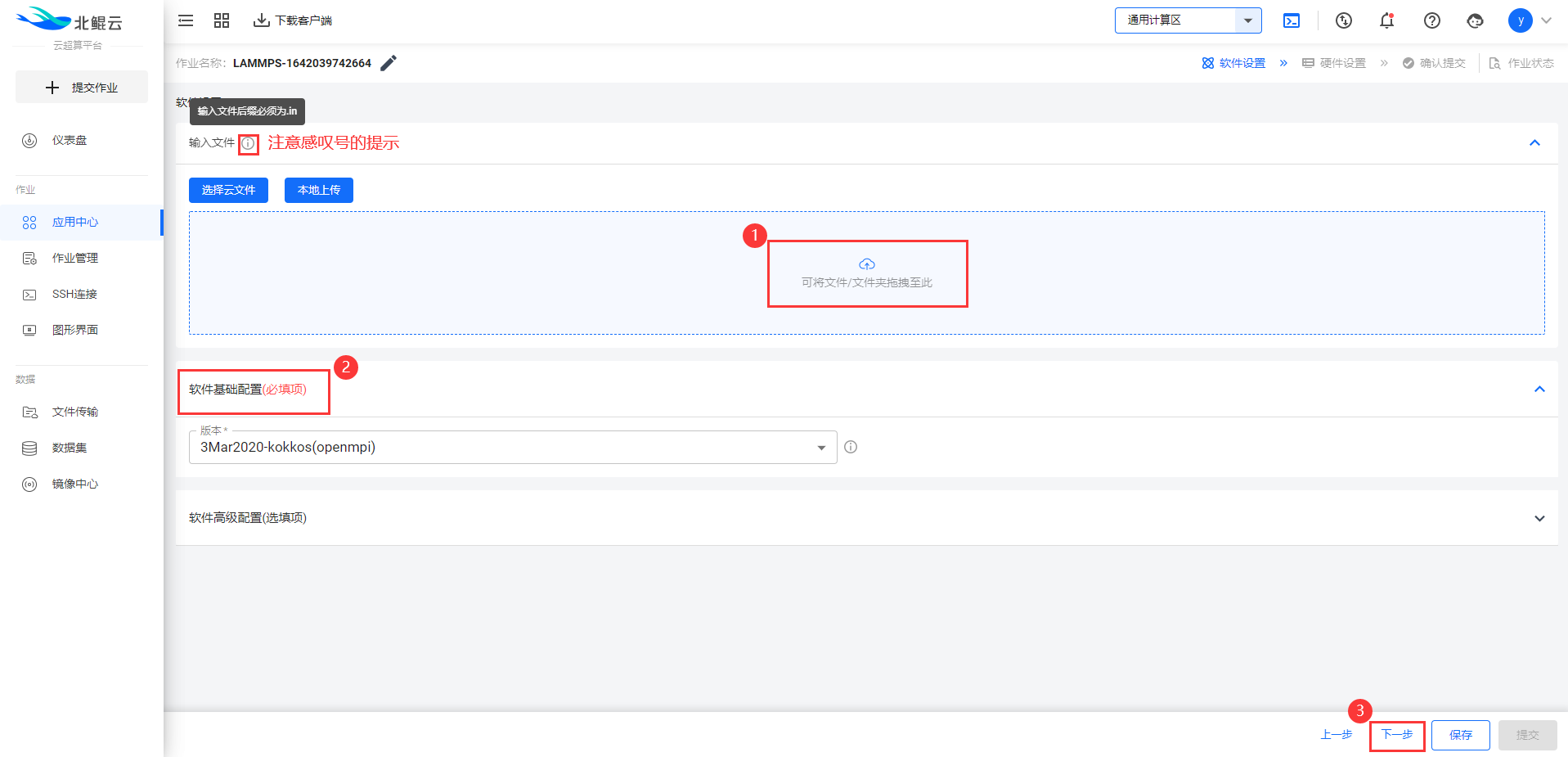
Step 4. 选择硬件配置;
节点数量:设置启动多少个并行计算的计算节点。
内存配比:设置各个计算节点内存大小为单节点核心数×内存配比。

Step 5. 查看作业内容汇总,并提交作业;

Step 6. 通过作业管理查看运行中的作业;
二. 命令行提交
通过SSH连接创建并连接管理节点。
Step 1. 创建作业目录并进入;
mkdir lammpsJob1
cd lammpsJob1
Step 2. 通过文件传输上传所需的输入文件,详情请查看Linux数据传输;
CPU版LAMMPS作业示例
Step 1. 在该文件夹下创建如下执行脚本lammps.sh:
#!/bin/bash
module add LAMMPS/23Jun2022-foss-2021a-kokkos
ulimit -s unlimited
ulimit -l unlimited
mpirun lmp -in M-1.in
Step 2. 使用slurm命令提交到计算节点;
2个4核心节点启动8个并行任务。
sbatch -N 2 -p c-4-2 -n 8 -c 1 lammps.sh
GPU版LAMMPS作业示例
Step 1. 在该文件夹下创建如下执行脚本lammps.sh:
#!/bin/bash
module add LAMMPS/lammps-2021.9-fosscuda.2019
ulimit -s unlimited
ulimit -l unlimited
lmp -sf gpu -pk gpu 0 neigh no -in M-1.in #输入文件
Step 2. 提交作业;
提交到单卡V100 GPU 节点。
sbatch -p g-v100-1 -c 10 lammps.sh
查看作业运行情况及参数详细介绍请点击查看slurm命令。
结果文件下载请查看Linux数据传输。
点击下载以上作业样例:LAMMPS.zip。