AlphaFold3
AlphaFold3 是由谷歌 DeepMind 和 Isomorphic Labs 团队开发的人工智能程序,于2024年11月开源供学术用途,它能准确预测蛋白质、DNA、RNA 等多种生物分子及其复合体的结构和相互作用,预测精度较 AlphaFold2 大幅提升,在药物研发等领域应用广泛。其架构引入 Pairformer 和扩散模块,采用跨蒸馏技术训练,减少了多重序列比对处理量,提高了计算效率和泛化能力,为生命科学研究提供了更强大的工具。
注意事项:
- AlphaFold3 仅供非商业用途使用,并受AlphaFold3 服务条款约束,如需使用集群上的 AlphaFold3,请填写 此表单 申请,权限将由 Google DeepMind 自行决定 。
- 运行AlphaFold3 作业需要选择单卡的GPU机型,多卡GPU没有加速效果。
- 对于长度过长、结构过于复杂的序列,受限于实际选择的GPU显卡性能和Alphafold3程序问题,不保证一定能输出正常的计算结果。
- 与 AlphaFold2不同, AlphaFold3 输入文件需要修改成json 格式。
单个蛋白输入文件示例:
{
"name": "2pv7",
"sequences": [
{
"protein": {
"id": ["A", "B"],
"sequence": "GMRESYANENQFGFKTINSDIHKIVIVGGYGKLGGLFARYLRASGYPISILDREDWAVAESILANADVVIVSVPINLTLETIERLKPYLTENMLLADLTSVKREPLAKMLEVHTGAVLGLHPMFGADIASMAKQVVVRCDGRFPERYEWLLEQIQIWGAKIYQTNATEHDHNMTYIQALRHFSTFANGLHLSKQPINLANLLALSSPIYRLELAMIGRLFAQDAELYADIIMDKSENLAVIETLKQTYDEALTFFENNDRQGFIDAFHKVRDWFGDYSEQFLKESRQLLQQANDLKQG"
}
}
],
"modelSeeds": [1],
"dialect": "alphafold3",
"version": 1
}
单个蛋白输入文件字段解析
顶层结构
- 描述: 包含与蛋白质序列和相关参数的建模任务相关的所有信息。
- 含义: 每个输入文件对应一个蛋白质或蛋白质复合物的建模任务,包含了蛋白质的序列、种子信息、模板、修改、外部 MSA 数据等。
name
- 类型: 字符串
- 示例值: "
2PV7" - 含义: 蛋白质或任务的名称/标识符。
- 用途: 用于唯一标识此建模任务,可以是具体的蛋白质名称、编号或任意标识符。
sequences
- 类型: 数组
- 示例值:
"sequences": [
{
"protein": {
"id": ["A", "B"],
"sequence": "GMRESYANENQFGFKTINSDIHKIVIVGGYGKLGGLFARYLRASGYPISILDREDWAVAESILANADVVIVSVPINLTLETIERLKPYLTENMLLADLTSVKREPLAKMLEVHTGAVLGLHPMFGADIASMAKQVVVRCDGRFPERYEWLLEQIQIWGAKIYQTNATEHDHNMTYIQALRHFSTFANGLHLSKQPINLANLLALSSPIYRLELAMIGRLFAQDAELYADIIMDKSENLAVIETLKQTYDEALTFFENNDRQGFIDAFHKVRDWFGDYSEQFLKESRQLLQQANDLKQG"
}
}
]
- 含义: 每个蛋白质的序列信息。每个蛋白体序列用一个对象表示,其中包括:
- id: 蛋白质链的标识符(如 "A" 或 "B")。
- sequence: 蛋白质链的氨基酸序列。
modelSeeds
- 类型: 数组(整数)
- 示例值: [1]
- 含义: 用于建模的种子数据,用来控制模型的初始化。这里为
[1]表示使用种子为 1 的模型。 - 用途: 提供多个随机种子值时,模型将基于这些种子生成多个可能的结构,以增加结果的可靠性或多样性。
dialect
- 类型: 字符串
- 示例值: "
alphafold3" - 含义: 指定输入文件的格式或方言。
- 用途: 确保模型理解并正确解析输入文件。在这个例子中,使用了 alphafold3 格式。
version
- 类型:整数
- 示例值:1
- 含义:表示输入文件的版本号,可以是1或2,1 是初始 AlphaFold 3 输入格式,2 是通过
unpairedMsaPath字段 和pairedMsaPath字段自定义指定外部的 MSA 文件,还有mmcifPath字段来指定外部自定义模板文件路径。
多聚体蛋白输入文件示例:
{
"name": "2PV7",
"sequences": [
{
"protein": {
"id": "A",
"sequence": "WNYLSEKRPNEPVHKHDQKAIITREWCPIFKSCLGFWIQGQGSKVLIYTFTIGSALKICYSAQHNNKNNANCGQSIAAEQNKMRFGLQTADRIIPTLYFEVQKPNEREQSCESLAFIYHFCVCPQGHAKKIHFHHMMNHGHTCWLALIHWLYWSSHTFRVQMFYWRPHVCNAQQCSDSSSPFLRRDTKMRQINYNMCYTFWYYDKCHEWMAWCTTHNPSYQVKCQQMYAAGHCWPPIELFFPGASQKEEPGHCCYYTLNAVVMKWQLETLDAMFGAQQLCVRSWASGFGPYEYFQDHPSRNYPKVHMLCIEANHKDRMADMDAEKKAKREIDGLSWEVMPREFHAGFWPRFFNVLHSEIWHSIWPELNRRRWLAKPWKECNFQWPWRDGKFWRQSFTWKKFIFWMCVDLVREFPMNERKSHHFAAVGRLPQRTYTAMLLPHWFNMHFCNTWPQFWDTWGESYKDFWFTKKPSVSRGDSYRISHTVPYFYIEFNKVFSDKM"
}
},
{
"protein": {
"id": "B",
"sequence": "SHHTPYQMHLFRSLAIYTHNYEHKDEIFEITVIAWLENIWGCRWLLFTMWMAHRISVAPPLRNGIYSHWPCHWEAGNPRLYRKIVQGEVIQPWHIHCCYPGDDINPGQCAMNDQAWMLFKWWPHVQAAVFKLSGYQEQNKQMGWSDHKPEWAQKRFFDYFRRHKTLGSPCIDVFIHQCNWVYYNAHHTWNLCISPYFYFPAMYFCDELMIHLVMVADFSNTNSAQLRHATIKSNKCKANHSNFIWITINVGMFKGWWIFCCRMVFKYGPNWNYLHHRMHVVAAVSSQDRMVCIMMNAFGRHRSDYRLSWAPDQVDGSGAWAEQEQTKCEGKMARWVRGTVIGANVAWCGPVYIYPDYECFAFAWWQGVEMRMYIIPKNVHMHQIVWFAPALACRMVCKCTPYQKHTAWDKYNHVHPIHYPYDRFIFTLFQPLSDCTFPSSAMSMWPRKPQDPMLIFMEMRMHTCDAQWCYNVREARIWDVVYGRPSHTEININMGVWKIE"
}
}
],
"modelSeeds": [1,2],
"dialect": "alphafold3",
"version": 1
}
多聚体蛋白输入文件字段解析
顶层结构
- 描述: 包含与蛋白质序列和相关参数的建模任务相关的所有信息。
- 含义: 每个输入文件对应一个蛋白质或蛋白质复合物的建模任务,包含了蛋白质的序列、种子信息、模板、修改、外部 MSA 数据等。
name
- 类型: 字符串
- 示例值: "
2PV7" - 含义: 蛋白质或任务的名称/标识符。
- 用途: 用于唯一标识此建模任务,可以是具体的蛋白质名称、编号或任意标识符。
sequences
- 类型: 数组
- 示例值:
"sequences": [
{
"protein": {
"id": "A",
"sequence": "WNYLSEKRPNEPVHKHDQKAIITREWCPIFKSCLGFWIQGQGSKVLIYTFTIGSALKICYSAQHNNKNNANCGQSIAAEQNKMRFGLQTADRIIPTLYFEVQKPNEREQSCESLAFIYHFCVCPQGHAKKIHFHHMMNHGHTCWLALIHWLYWSSHTFRVQMFYWRPHVCNAQQCSDSSSPFLRRDTKMRQINYNMCYTFWYYDKCHEWMAWCTTHNPSYQVKCQQMYAAGHCWPPIELFFPGASQKEEPGHCCYYTLNAVVMKWQLETLDAMFGAQQLCVRSWASGFGPYEYFQDHPSRNYPKVHMLCIEANHKDRMADMDAEKKAKREIDGLSWEVMPREFHAGFWPRFFNVLHSEIWHSIWPELNRRRWLAKPWKECNFQWPWRDGKFWRQSFTWKKFIFWMCVDLVREFPMNERKSHHFAAVGRLPQRTYTAMLLPHWFNMHFCNTWPQFWDTWGESYKDFWFTKKPSVSRGDSYRISHTVPYFYIEFNKVFSDKM"
}
},
{
"protein": {
"id": "B",
"sequence": "SHHTPYQMHLFRSLAIYTHNYEHKDEIFEITVIAWLENIWGCRWLLFTMWMAHRISVAPPLRNGIYSHWPCHWEAGNPRLYRKIVQGEVIQPWHIHCCYPGDDINPGQCAMNDQAWMLFKWWPHVQAAVFKLSGYQEQNKQMGWSDHKPEWAQKRFFDYFRRHKTLGSPCIDVFIHQCNWVYYNAHHTWNLCISPYFYFPAMYFCDELMIHLVMVADFSNTNSAQLRHATIKSNKCKANHSNFIWITINVGMFKGWWIFCCRMVFKYGPNWNYLHHRMHVVAAVSSQDRMVCIMMNAFGRHRSDYRLSWAPDQVDGSGAWAEQEQTKCEGKMARWVRGTVIGANVAWCGPVYIYPDYECFAFAWWQGVEMRMYIIPKNVHMHQIVWFAPALACRMVCKCTPYQKHTAWDKYNHVHPIHYPYDRFIFTLFQPLSDCTFPSSAMSMWPRKPQDPMLIFMEMRMHTCDAQWCYNVREARIWDVVYGRPSHTEININMGVWKIE"
}
}
]
- 含义: 每个蛋白质的序列信息。每个蛋白体序列用一个对象表示,其中包括:
- id: 蛋白质链的标识符(如 "A" 或 "B")。
- sequence: 蛋白质链的氨基酸序列。
modelSeeds
- 类型: 数组(整数)
- 示例值: [1, 2]
- 含义: 指定随机种子,以便模型生成多个预测结果。
- 用途: 提供多个随机种子值时,模型将基于这些种子生成多个可能的结构,以增加结果的可靠性或多样性。
dialect
- 类型: 字符串
- 示例值: "
alphafold3" - 含义: 指定输入文件的格式或方言。
- 用途: 确保模型理解并正确解析输入文件。在这个例子中,使用了 alphafold3 格式。
version
- 类型:整数
- 示例值:1
- 含义:表示输入文件的版本号,可以是1或2,1 是初始 AlphaFold 3 输入格式,2 是通过
unpairedMsaPath字段 和pairedMsaPath字段自定义指定外部的 MSA 文件,还有mmcifPath字段来指定外部自定义模板文件路径。
RNA输入文件示例:
{
"name": "rna",
"sequences": [
{
"rna": {
"id": ["A"],
"sequence": "AGCU",
"modifications": [
{"modificationType": "2MG","basePosition": 1},
{"modificationType": "5MC","basePosition": 4}
]
}
}
],
"modelSeeds": [1],
"dialect": "alphafold3",
"version": 1
}
RNA 输入文件字段解析
顶层字段:rna
- 类型:对象
- 用途:指定单个 RNA 链的相关信息,包括序列、修饰、以及多序列比对(MSA)的内容。
内部字段解析:
id
- 类型:
str或list[str] - 示例值:
"A"或["A", "B", "C"] - 含义:RNA 链的唯一标识符。
- 如果是字符串,表示单个 RNA 链的 ID。
- 如果是字符串列表,表示多个相同 RNA 链(同源链)的拷贝。
- 用途:在输出的 mmCIF 文件中使用该 ID 作为标识。
sequence
- 类型:
str - 示例值:
"AGCU" - 含义:RNA 链的核苷酸序列。
- 使用
A、C、G、U表示序列中的碱基。
- 使用
- 用途:定义 RNA 链的主要结构信息。
modifications
- 类型:
list[RnaModification](可选) - 示例值:
[
{"modificationType": "2MG", "basePosition": 1},
{"modificationType": "5MC", "basePosition": 4}
]
- 含义:RNA 链上的修饰信息。
modificationType:修饰类型,使用 CCD 代码(化学组件字典代码)表示。basePosition:修饰所在碱基的位置,1 为基准的索引。
- 用途:描述 RNA 链中碱基的化学修饰,影响功能或结构。
unpairedMsa
- 类型:
str(可选) - 示例值:A3M 格式的多序列比对内容。
- 含义:该链的可选多序列比对(MSA)信息,内联提供。
- 用途:为链提供多序列比对的背景信息以支持建模。
unpairedMsaPath
- 类型:
str(可选) - 示例值:
"/path/to/msa.a3m" - 含义:多序列比对文件的路径。
- 路径可以是绝对路径或相对于输入 JSON 文件的相对路径。
- 文件必须为 A3M 格式,可以是纯文本或压缩格式(gzip、xz、zstd)。
- 用途:与
unpairedMsa互斥,用于外部文件提供 MSA 数据。
modelSeeds
- 类型: 数组(整数)
- 示例值: [1, 2]
- 含义: 指定随机种子,以便模型生成多个预测结果。
- 用途: 提供多个随机种子值时,模型将基于这些种子生成多个可能的结构,以增加结果的可靠性或多样性。
dialect
- 类型: 字符串
- 示例值: "
alphafold3" - 含义: 指定输入文件的格式或方言。
- 用途: 确保模型理解并正确解析输入文件。在这个例子中,使用了 alphafold3 格式。
version
- 类型:整数
- 示例值:1
- 含义:表示输入文件的版本号,可以是1或2,1 是初始 AlphaFold 3 输入格式,2 是通过
unpairedMsaPath字段 和pairedMsaPath字段自定义指定外部的 MSA 文件,还有mmcifPath字段来指定外部自定义模板文件路径。
DNA输入文件示例:
{
"name": "dna",
"sequences": [
{
"dna": {
"id": ["A"],
"sequence": "GACCTCT",
"modifications": [
{"modificationType": "6OG","basePosition": 1},
{"modificationType": "6MA","basePosition": 2}
]
}
}
],
"modelSeeds": [1],
"dialect": "alphafold3",
"version": 1
}
DNA 输入文件字段解析
顶层字段:dna
- 类型:对象
- 用途:描述 DNA 链的相关信息,包括其 ID、序列以及可选的修饰。
内部字段
id
- 类型:字符串或字符串数组 (
str | list[str]) - 示例值:
"A"或["A", "B", "C"] - 含义:
- 唯一标识 DNA 链的标识符。
- 当指定为单个字符串(如
"A")时,表示只有一个 DNA 链。 - 当指定为一个字符串数组(如
["A", "B", "C"])时,表示同构链的多个拷贝。
- 用途:在输出文件(如 mmCIF 文件)中也会使用这些 ID。
- 类型:字符串或字符串数组 (
sequence
- 类型:字符串
- 示例值:
"GACCTCT" - 含义:DNA 的序列,由碱基的单字母代码表示,包括
A(腺嘌呤)、C(胞嘧啶)、G(鸟嘌呤)和T(胸腺嘧啶)。 - 用途:用于建模的基础数据,定义 DNA 链的化学结构。
modifications
- 类型:列表 (
list[DnaModification]) - 示例值:
[
{"modificationType": "6OG", "basePosition": 1},
{"modificationType": "6MA", "basePosition": 2}
] - 含义:描述 DNA 链中碱基的化学修饰。
- modificationType:字符串,表示修饰类型,通常用 CCD 代码标识(例如,
"6OG"表示氧化鸟嘌呤,"6MA"表示甲基腺嘌呤)。 - basePosition:整数,表示修饰位置,基于 1 的索引。
- modificationType:字符串,表示修饰类型,通常用 CCD 代码标识(例如,
- 用途:
- 定义 DNA 链中特定位置的化学修饰。
- 在模拟中用来考虑 DNA 的非标准化学性质。
- 类型:列表 (
modelSeeds
- 类型: 数组(整数)
- 示例值: [1, 2]
- 含义: 指定随机种子,以便模型生成多个预测结果。
- 用途: 提供多个随机种子值时,模型将基于这些种子生成多个可能的结构,以增加结果的可靠性或多样性。
dialect
- 类型: 字符串
- 示例值: "
alphafold3" - 含义: 指定输入文件的格式或方言。
- 用途: 确保模型理解并正确解析输入文件。在这个例子中,使用了 alphafold3 格式。
version
- 类型:整数
- 示例值:1
- 含义:表示输入文件的版本号,可以是1或2,1 是初始 AlphaFold 3 输入格式,2 是通过
unpairedMsaPath字段 和pairedMsaPath字段自定义指定外部的 MSA 文件,还有mmcifPath字段来指定外部自定义模板文件路径。
一. 命令行提交
通过SSH连接创建并连接管理节点。
Step 1. 创建工作目录并进入,也同时创建input 目录,在 input 目录中放置输入 JSON 文件,自定义输入 JSON 文件可参考官方文档 https://github.com/google-deepmind/alphafold3/blob/main/docs/input.md
mkdir -p $HOME/alphafold3Job1/input
cd $HOME/alphafold3Job1
Step 2. 通过文件传输上传所需输入文件fold_input.json,详情请查看Linux数据传输;
Step 3. 在该文件夹下创建如下执行脚本run_alphafold3.sh:
run_alphafold.py 可以通过以下两种方式之一提供输入:
- 单个输入文件:使用
--json_path标志后跟单个 JSON 文件的路径。- 多个输入文件:使用
--input_dir标志,后跟 JSON 文件目录的路径。--jackhmmer_n_cpu=16参数用于指定运行 Jackhmmer 工具搜索序列数据库使用的 CPU 核心数--nhmmer_n_cpu=16参数用于指定运行 Nhmmer 工具搜索RNA序列数据库使用的 CPU 核心数--json_path=/tmp/fold_input.json指定输入文件名称,需要修改成自己的输入文件名称,路径无需更改,如果是批量提交JSON 文件,指定JSON输入文件目录的路径,更改成--input_dir参数
vim run_alphafold3.sh
#!/bin/bash
export PROGRAM=/public/software/.local/easybuild/software/alphafold
# 创建输出目录和添加权限
mkdir -p $HOME/alphafold3Job1/output
chmod -R 777 $HOME/alphafold3Job1/output
cd $PROGRAM/alphafold3 && docker load < alphafold.tar
docker run -i -e XLA_FLAGS=--xla_disable_hlo_passes=custom-kernel-fusion-rewriter --privileged \
--volume $HOME/alphafold3Job1/input:/tmp/af_input \
--volume $HOME/alphafold3Job1/output:/tmp/af_output \
--volume $PROGRAM/alphafold3-models:/root/models \
--volume $PROGRAM/alphafold3-databases/v3.0:/root/public_databases \
--gpus all \
alphafold3 \
python run_alphafold.py \
--jackhmmer_n_cpu=16 \
--nhmmer_n_cpu=16 \
--flash_attention_implementation=xla \
--json_path=/tmp/af_input/fold_input.json \
--model_dir=/root/models \
--output_dir=/tmp/af_output
step 4. 给脚本添加执行权限
chmod +x run_alphafold3.sh
Step 5. 提交作业;
提交任务到带有一张A10显卡的GPU节点运行。
sbatch -p g-a10-1 -c 16 run_alphafold3.sh
查看作业运行情况及参数详细介绍请点击查看slurm命令。
结果文件下载请查看Linux数据传输。
二. CPU和GPU分步骤计算(可选)
说明:因为alphafold3计算周期整个过程中,前面三分之二时间是通过Jackhmmer 工具或者Nhmmer 工具搜索数据库,使用消耗的是CPU 算力,根据用户需求,此部分可根据需求选择,如嫌弃麻烦,可全程使用gpu节点提交任务计算,因部分用户预算有限,需要批量提交多条蛋白计算任务,节约计算成本,缩短计算时长,从而需要分开计算,可选择CPU和GPU分步骤计算方案。
参数说明:
--comment disk:CLOUD_ESSD:40g指定申请计算节点的磁盘大小,cpu计算节点默认20g,导入docker 计算环境镜像存储空间不够,需要增加到40g。
--dependency=afterok:$cpu_job_id提交GPU计算步骤时,确保 GPU 任务只会在 CPU 筛选任务成功完成后才会启动,$cpu_job_id 是指前面步骤cpu完成的任务id,如下述案例中,查询提交计算成功的任务id 为 6。
一. CPU 计算部分
通过SSH连接创建并连接管理节点。
Step 1. 创建工作目录并进入,也同时创建input 目录,在 input 目录中放置输入 JSON 文件,自定义输入 JSON 文件可参考官方文档 https://github.com/google-deepmind/alphafold3/blob/main/docs/input.md
mkdir -p $HOME/alphafold3Job1/input
cd $HOME/alphafold3Job1
Step 2. 通过北鲲云控制台文件传输,上传所需输入文件fold_input.json,详情请查看Linux数据传输;
Step 3. 在该文件夹下创建如下执行脚本run_alphafold3_cpu.sh:
vim run_alphafold3_cpu.sh
#!/bin/bash
export PROGRAM=/public/software/.local/easybuild/software/alphafold
# 创建输出目录和添加权限
mkdir -p $HOME/alphafold3Job1/output
chmod -R 777 $HOME/alphafold3Job1/output
cd $PROGRAM/alphafold3 && docker load < alphafold.tar
docker run -i --privileged \
--volume $HOME/alphafold3Job1/input:/tmp/af_input \
--volume $HOME/alphafold3Job1/output:/tmp/af_output \
--volume $PROGRAM/alphafold3-models:/root/models \
--volume $PROGRAM/alphafold3-databases/v3.0:/root/public_databases \
alphafold3 \
python run_alphafold.py \
--jackhmmer_n_cpu=64 \
--nhmmer_n_cpu=64 \
--flash_attention_implementation=xla \
--json_path=/tmp/af_input/fold_input.json \
--model_dir=/root/models \
--output_dir=/tmp/af_output \
--run_inference=false
step 4. 给脚本添加执行权限
chmod +x run_alphafold3_cpu.sh
Step 5. 提交作业;
提交任务到一台64核心的CPU节点上运行。
sbatch -p c-64-1 -c 64 --comment disk:CLOUD_ESSD:80g run_alphafold3_cpu.sh
二. GPU 计算部分
注意:CPU步骤算完之后,需要要把output里的新json文件拿出来作为GPU计算部分的输入,CPU计算部分的输入文件需要删除
Step 1. 进入CPU部分计算完成的作业任务文件夹;
cd $HOME/alphafold3Job1
Step 2. 在该文件夹下创建如下执行脚本run_alphafold3_gpu.sh:
vim run_alphafold3_gpu.sh
#!/bin/bash
export PROGRAM=/public/software/.local/easybuild/software/alphafold
# 创建输出目录和添加权限
mkdir -p $HOME/alphafold3Job1/output
chmod -R 777 $HOME/alphafold3Job1/output
cd $PROGRAM/alphafold3 && docker load < alphafold.tar
docker run -i --privileged \
--volume $HOME/alphafold3Job1/input:/tmp/af_input \
--volume $HOME/alphafold3Job1/output:/tmp/af_output \
--volume $PROGRAM/alphafold3-models:/root/models \
--volume $PROGRAM/alphafold3-databases/v3.0:/root/public_databases \
--gpus all \
alphafold3 \
python run_alphafold.py \
--json_path=/tmp/af_input/fold_input_data.json \
--model_dir=/root/models \
--output_dir=/tmp/af_output
--run_data_pipeline=false
step 3. 给脚本添加执行权限
chmod +x run_alphafold3_gpu.sh
Step 4. 提交作业;
提交任务到带有一张A10显卡的GPU节点上运行,并指定CPU提交计算的任务ID。
查询所有最近的历史任务
这个命令会列出最近一天内的所有作业,包括任务 ID(JobID)、任务名称(JobName)、分区(Partition)、任务状态(State)和退出码(ExitCode)。
sacct --format=JobID,JobName,Partition,State,ExitCode -S $(date -d '-1 days' +%Y-%m-%d)
sbatch -p g-a10-1 -c 16 --dependency=afterok:6 run_alphafold3_gpu.sh
查看作业运行情况及参数详细介绍请点击查看slurm命令。
结果文件下载请查看Linux数据传输。
三. 结果文件介绍
正常的计算结果应包含有以下文件结构:
下面是一个名为“2pv7”的 AlphaFold 3 输出目录列表示例,该作业已使用 2 个种子和 2 个样本运行
output/2pv7
├── 2pv7_confidences.json
├── 2pv7_data.json
├── 2pv7_model.cif
├── 2pv7_summary_confidences.json
├── ranking_scores.csv
├── seed-1_sample-0
│ ├── confidences.json
│ ├── model.cif
│ └── summary_confidences.json
├── seed-1_sample-1
│ ├── confidences.json
│ ├── model.cif
│ └── summary_confidences.json
├── seed-1_sample-2
│ ├── confidences.json
│ ├── model.cif
│ └── summary_confidences.json
├── seed-1_sample-3
│ ├── confidences.json
│ ├── model.cif
│ └── summary_confidences.json
├── seed-1_sample-4
│ ├── confidences.json
│ ├── model.cif
│ └── summary_confidences.json
├── seed-2_sample-0
│ ├── confidences.json
│ ├── model.cif
│ └── summary_confidences.json
├── seed-2_sample-1
│ ├── confidences.json
│ ├── model.cif
│ └── summary_confidences.json
├── seed-2_sample-2
│ ├── confidences.json
│ ├── model.cif
│ └── summary_confidences.json
├── seed-2_sample-3
│ ├── confidences.json
│ ├── model.cif
│ └── summary_confidences.json
├── seed-2_sample-4
│ ├── confidences.json
│ ├── model.cif
│ └── summary_confidences.json
└── TERMS_OF_USE.md
四. 置信度指标
与 AlphaFold2 和 AlphaFold-Multimer 类似,AlphaFold 3 输出包括置信度指标。
pLDDT:
- 0-100 范围内的每个原子置信度估计,值越高表示置信度越高。
- pLDDT 旨在预测仅考虑聚合物距离的改进的 LDDT 分数。
- 对于蛋白质,这类似于 lDDT-Cα 度量,但粒度更大,因为它可以因原子而异,而不仅仅是因残基而异。
- 对于配体原子,改进的 LDDT 仅考虑配体原子和聚合物之间的误差,而不考虑其他配体原子。
- 对于 DNA/RNA,改进的 LDDT 使用更宽的半径 30 Å,而不是 15 Å。
PAE(预测对齐误差):
- 预测结构中两个标记之间的相对位置和方向的误差估计值值越高表示预测误差越大,因此置信度越低。
- 对于蛋白质和核酸,PAE 分数与 AlphaFold2 基本相同,其中误差是相对于由蛋白质骨架构建的框架来测量的。
- 对于小分子和翻译后修饰,每个原子的框架都是从参考构象异构体的最近邻居构建的。
pTM 和 ipTM 分数:
- 预测模板建模 (pTM) 分数和界面预测模板建模 (ipTM) 分数均来自称为模板建模 (TM) 分数的度量。
- 这可衡量整个结构的准确性(Zhang and Skolnick, 2004; Xu and Zhang, 2010)。
- pTM 分数高于 0.5 表示预测的复合物整体折叠可能与真实结构相似。
- ipTM 可衡量复合物内亚基相对位置预测的准确性。
- 高于 0.8 的值代表有信心的高质量预测,而低于 0.6 的值则表示预测失败。
- 0.6 到 0.8 之间的 ipTM 值是灰色区域,
- 预测可能正确也可能不正确。
- TM 分数对于小结构或短链非常严格,因此当涉及的标记少于 20 个时,pTM 会分配小于 0.05 的值;对于这些情况,PAE 或 pLDDT 可能更能表明预测质量。
- AlphaFold3 GitHub 官方输出文件目录结构和可信度说明: https://github.com/google-deepmind/alphafold3/blob/main/docs/output.md
- AlphaFold 3 论文: https://www.nature.com/articles/s41586-024-07487-w
五. 使用PyMOL对结果进行图形化展示
Step 1. 应用中心搜索PyMOL;
Step 2. 点击提交作业,选择图形界面提交;
Step 3. 启动PyMOL,选择硬件配置并启动,通过VNC连接启动的linux工作站,使用软件;
Step 4. 点击File 找到结果文件输出路径,打开.cif结尾的文件,查看输出;

六. 使用在线工具PAE Viewer 展示结构图和矢量图
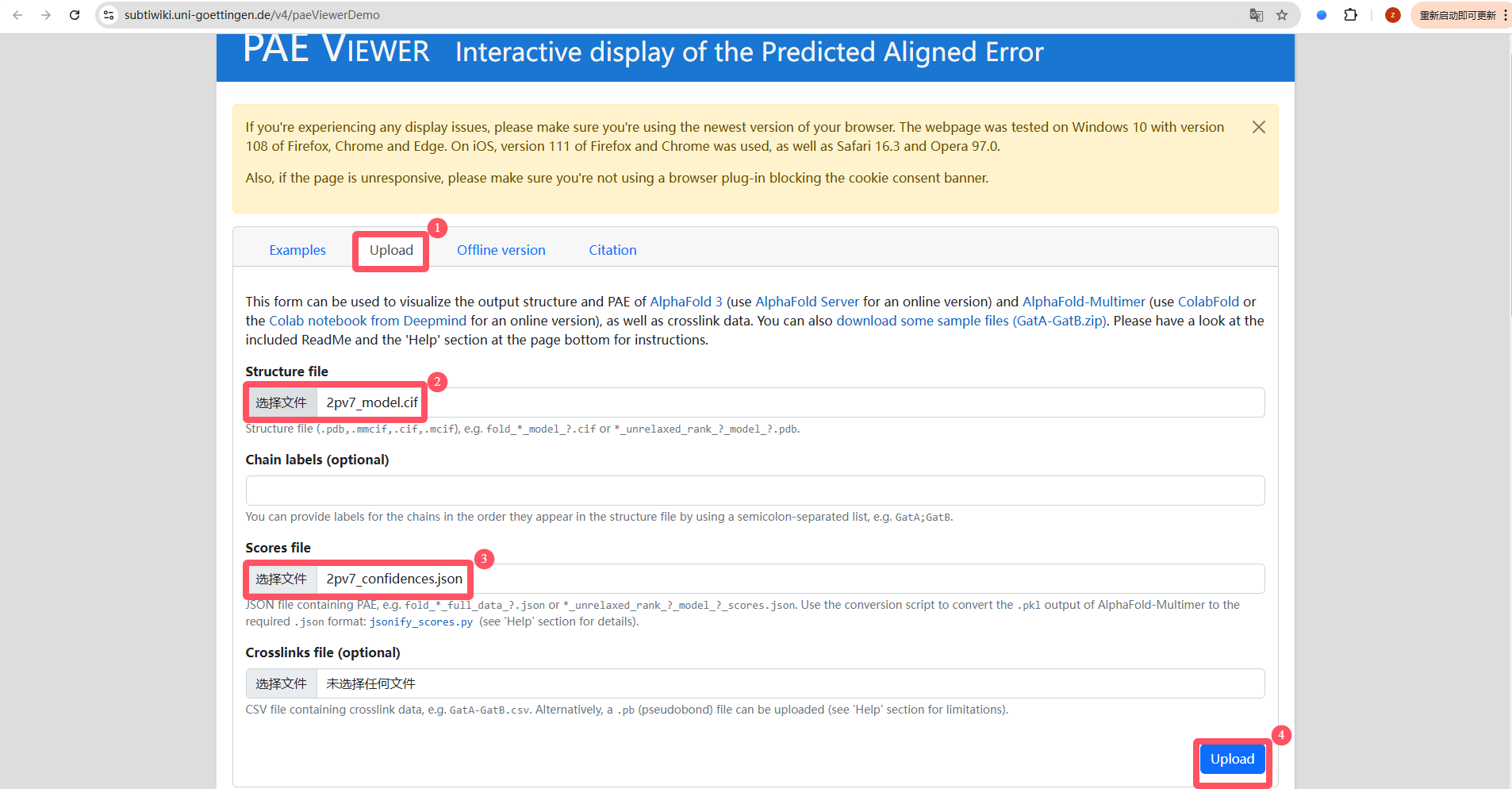
Step 1. 在浏览器中输入:https://subtiwiki.uni-goettingen.de/v4/paeViewerDemo
Step 2. 上传结果文件中2pv7_model.cif和2pv7_confidences.json文件,点击upload按钮;
Step 3. 展示计算生成的结构图和矢量图。
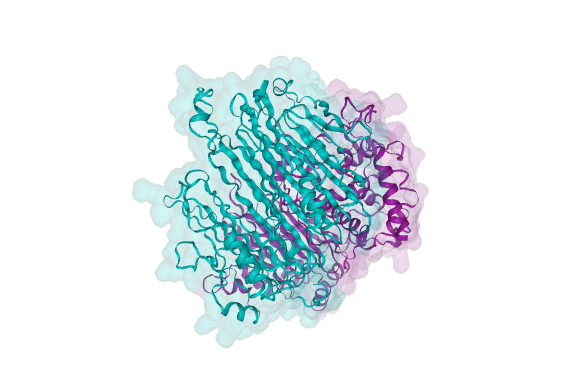 |  |
|---|---|
| 结构 图 | 矢量 图 |