HelixFold-Single
HelixFold-Single从近3亿的无标注蛋白质数据中提取信息,建模蛋白质之间的关系,从而将MSA同源信息隐式的学习在预训练大模型中,进而有效地替代MSA信息检索模块,极大地提升了结构预测的速度,模型推理的速度平均提升数百倍。
注意事项:
- 运行HelixFold-Single 作业,只能选择GPU 计算区,V100或者A100 单卡资源进行计算。
- 输入文件名不能有特殊符号和
空格。 - 提交的文件第一行为标题,第二行为序列,序列为大写字母,序列之间不能有
空格或换行符。 - 模板提交一次上传多个序列文件,会启动多个计算节点,每个计算节点对应一个序列文件。
单体结构输入文件示例:
>sequence_1
GSDNGFGSSKATSGSDFGGLAIFDGSGSEHFGHSDTHGSFDGLFGVDFZILSQQLKS
一. 模板提交
Helixfold-Single 使用模板提交,会根据上传的输入文件个数,启动对应数量的计算节点。如:上传5个输入文件,硬件配置选择节点数量为1提交作业,默认会启动5个作业节点同时计算,相应的以5个节点的价格进行计费;相比于串行提交,以相同的成本节约了计算的时间。
Step 1. 在应用中心搜索Helixfold-Single软件,点击提交作业-选择模板提交;

Step 2.选择可视化模板提交;

Step 3. 上传序列文件(.fasta格式)

Step 4. 选择GPU硬件配置虎鲸1卡(v100-1);

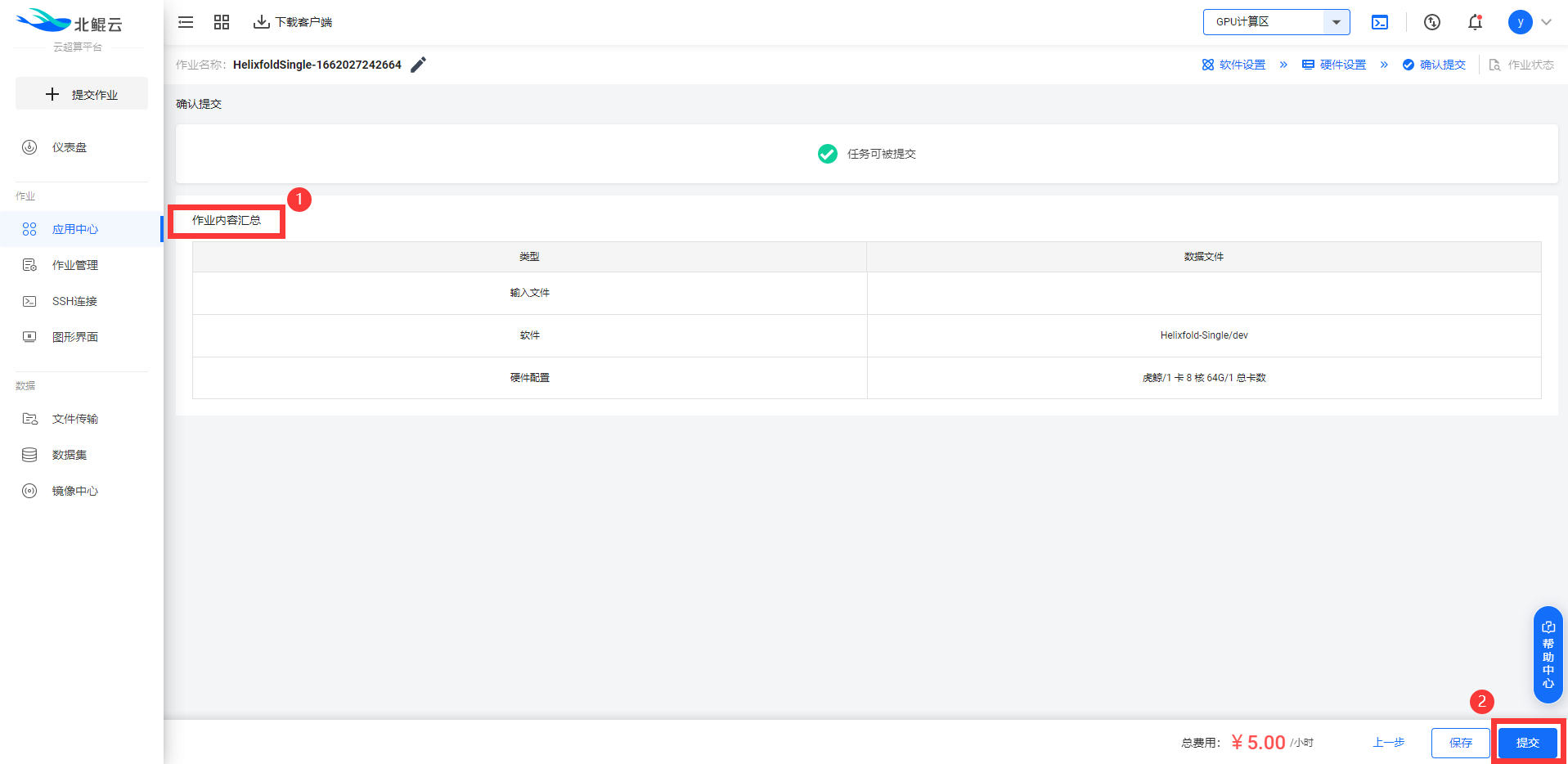
Step 5. 查看作业内容汇总,并提交作业;
Step 6. 通过作业管理查看运行中的作业;
二. 命令行提交
通过SSH连接创建并连接管理节点。
Step 1. 创建作业目录并进入;
mkdir Helixfold-SingleJob1
cd Helixfold-SingleJob1
Step 2. 通过文件传输上传所需的输入文件test.fasta,详情请查看Linux数据传输;
Step 3. 在该文件夹下创建如下执行脚本Helixfold-Single.sh:
#!/bin/bash
module add Anaconda3/2020.02
source activate helixfold-single
export CUDA_VISIBLE_DEVICES=0
export LD_LIBRARY_PATH=/public/software/.local/easybuild/software/Anaconda3/2020.02/envs/helixfold-single/lib
ROOT_PATH="$(pwd)"
init_model="/public/software/.local/easybuild/software/helixfold-single/helixfold-single.pdparams"
######## Predict protein by HelixFold-single.
python /public/software/.local/easybuild/software/helixfold-single/helixfold_single_inference.py \
--init_model=${init_model} \
--fasta_file=$ROOT_PATH/test.fasta \
--output_dir=$ROOT_PATH \
--num_outputs=5
Step 4. 提交作业;
提交任务到带有一张V100卡的GPU节点运行。
sbatch -p g-v100-1 -c 8 Helixfold-Single.sh
查看作业运行情况及参数详细介绍请点击查看slurm命令。
结果文件下载请查看Linux数据传输。
三. HelixFold-Single介绍
PR:https://www.paddlepaddle.org.cn/support/news?action=detail&id=3033
GitHub地址:https://github.com/PaddlePaddle/PaddleHelix/tree/dev/apps/protein_folding/helixfold
论文地址:https://arxiv.org/abs/2207.13921
四. 使用PyMOL对结果进行图形化展示
Step 1. 应用中心搜索PyMOL;
Step 2. 点击提交作业,选择图形界面提交;
Step 3. 启动PyMOL,选择硬件配置并启动;
Step 4. 通过VNC连接启动的linux工作站,使用软件;