


此前,何博士为大家在北鲲云的直播间分享了Amber热力学积分计算相对自由能变化(直播回放可在视频号:北鲲云-直播回放中查看)
直播结束后有很多小伙伴来向我们要PPT资料,这里何博士也为大家准备了文字版本的教程。将为大家介绍如何在北鲲云计算平台上利用Amber热力学积分计算相对自由能变化,体系包括小分子-蛋白(小分子改变),小分子-蛋白(蛋白突变),蛋白-蛋白相互作用。
本教程要求使用者一定程度了解Amber动力学模拟程序。
Amber是美国加州大学Peter Kollman等开发的一款著名的分子动力学模拟软件包。Amber主要适用于蛋白质,小分子和多糖等生物分子体系的模拟。
本文所需的所有文件请在https://github.com/Xinheng-He/ti_toturial上下载。

该应用场景解决将蛋白口袋内的小分子A变为小分子B所产生的相对自由能变。
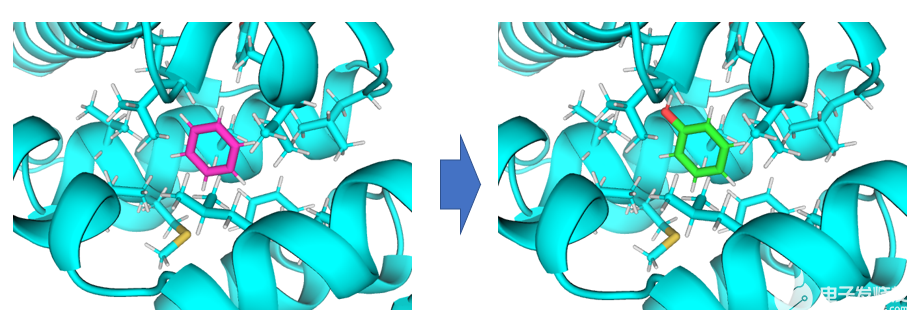
将蛋白口袋内的苯转化为苯酚。
首先,使用pymol将分子打开,并选中小分子,保存为mol2文件,如下图所示,我们使用
save *my_case\ben_ligand.mol2
save *\my_case\benfen_ligand.mol2
这两个命令保存了变化前后的配体。
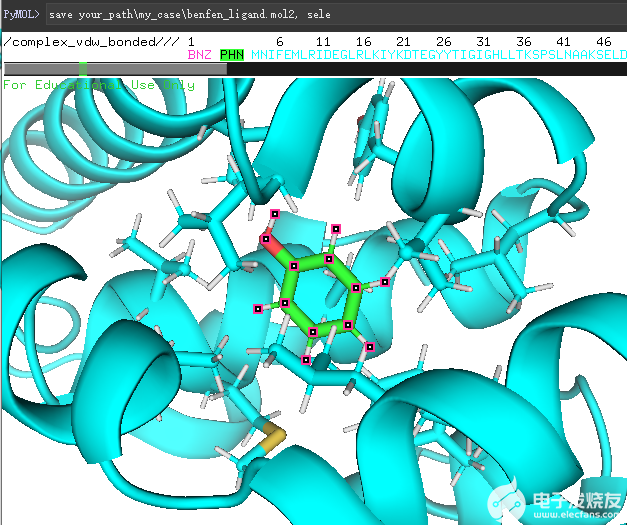
(保存pymol中的sele对象)
开启一个北鲲云管理节点加载环境。
module add Anaconda3/2020.02
source/public/software/.local/easybuild/software/amber/aber20/amber.sh
ulimit-s unlimited
ulimit-l unlimited
对苯生成具有电荷的可用mol2文件,总电荷为0,残基名为BEN。
antechamber-i ben_ligand.mol2 -fi mol2 -o ben_real.gaff2.mol2 -fo mol2
生成frcmod力场参数文件。
parmchk2-i ben_real.gaff2.mol2 -f mol2 -o BEN.gaff2.frcmod-s gaff2-a yes
上述操作对苯酚再来一次。
antechamber -i benfen_ligand.mol2 -fi mol2 -o benfen_real.gaff2.mol2 -fo mol2 -rn FEN-atgaff2-anyes-drno-pfyes-cbcc-nc0
parmchk2 -i benfen_real.gaff2.mol2 -f mol2 -o FEN.gaff2.frcmod -s gaff2 -a yes
根据frcmod文件,生成两个分子的文库文件,该文件描述了分子内部的原子类型和键连信息。
tleap-f-<<_EOF
source leaprc.gaff2
loadamberparams FEN.gaff2.frcmod
FEN = loadmol2 benfen_real.gaff2.mol2
saveoff FEN FEN.lib
savepdb FEN FEN.pdb
quit
_EOF
tleap -f - <<_EOF
source leaprc.gaff2
loadamberparams BEN.gaff2.frcmod
BEN = loadmol2 ben_real.gaff2.mol2
saveoff BEN BEN.lib
savepdb BEN BEN.pdb
quit
_EOF
注意前后的力场要保持一致。
将两个pdb文件(FEN.pdb和BEN.pdb)中同样位置的全部重原子,保存成同样的坐标,注意名字要和lib中的一样,放成一个lig.pdb,在下面的tleap过程中,tleap会自动根据lib文件将complex中的原子变成真实的样子,这样做是为了保证一样原子的位置完全一致,减少不必要的变量。
(pdb文件的内容)
使用pdb4amber,检查蛋白是否有二硫键,或需要编辑的残基。
pdb4amber pure_protein.pdb -o pure_check.pdb
cat pure_check_sslink
没有二硫键,之后使用pure_check.pdb。
接下来在tleap中加载配体和受体。
tleap -f - <<_EOF
source leaprc.protein.ff14SB
source leaprc.gaff2
source leaprc.water.tip3p
loadAmberParams frcmod.ionsjc_tip3p
loadoff BEN.lib
loadoff FEN.lib
loadamberparams BEN.gaff2.frcmod
loadamberparams FEN.gaff2.frcmod
ligands = loadpdb lig.pdb
complex = loadpdb pure_check.pdb
complex = combine {ligands complex}
check complex
solvatebox ligands TIP3PBOX 15
addions ligands Na+ 0
savepdb ligands ligands_vdw_bonded.pdb
saveamberparm ligands ligands_vdw_bonded.parm7 ligands_vdw_bonded.rst7
solvatebox complex TIP3PBOX 15
addions complex Cl- 0
savepdb complex complex_vdw_bonded.pdb
saveamberparm complex complex_vdw_bonded.parm7 complex_vdw_bonded.rst7
quit
_EOF
注意根据实际电荷情况调整addions,如果ligand/complex带负电,加Na+,反之加Cl-,离子类型可以自己选择。
下载complex_vdw_bonded.pdb并检查其中的配体分子区别,是否只是不一样的配体部分不一样,确定配体不一样部分的原子。设置出发和结束原子。
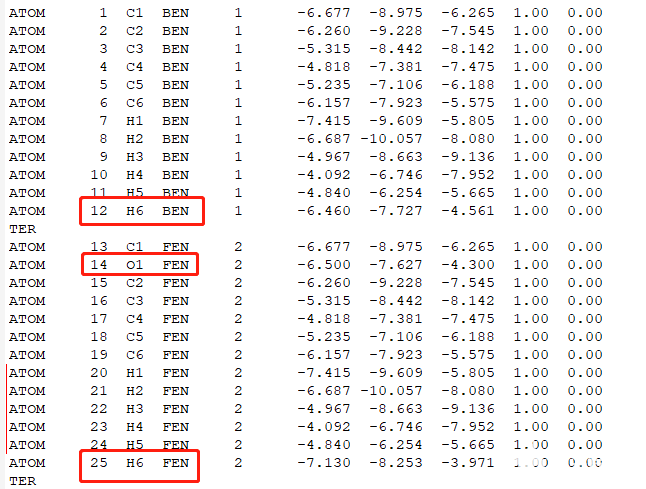
红框是有区别的原子,其他原子需要手动编辑使其保持一致的坐标,红线是编辑后的原子。
修改initial中的in文件下述部分。
timask1=':BEN',timask2=':FEN',
scmask1=':BEN@H6',scmask2=':FEN@O1,H6'
使6个in文件符合实际更改的原子情况,只改变不同的原子,该步骤的目标是优化vdw变化过程中氢原子的位置。
使用sbatch run_v100.slurm,将任务提交到北鲲云的单卡V100集群上,该任务大概耗时1分钟,注意该任务如果有报错,可能是以下问题:
如果上述过程报关于ambmask的错(比如mask中不能用_符号),可以改写ambmask来获得正确可识别的mask。
ambmask-p complex_vdw_bonded.parm7 -c complex_vdw_bonded.rst7 -find :FEN
是ambmask的输入方式,在parm7和rst7中寻找这些原子,并用pdb的形式输出。
如果报错Error : Atom 12 does not have match in V1 ,说明这个原子在两个小分子配体中间的位置区别太大,TI不能识别这两个分子作为一样的背景,因此将这两个原子(初始和结束配体的)加入坐标中,就可以解决这个问题。
运行结束后,提取优化过后的分子结构。
p ligands_vdw_bonded.rst7 ligands_vdw_bonded.rst7.leap
cp press_lig.rst7 ligands_vdw_bonded.rst7
cp complex_vdw_bonded.rst7 complex_vdw_bonded.rst7.leap
cp press_com.rst7 complex_vdw_bonded.rst7
cpptraj -p ligands_vdw_bonded.parm7 <<_EOF
trajin ligands_vdw_bonded.rst7
strip ":1,2"
outtraj ligands_solvated.pdb onlyframes 1
unstrip
strip ":2-999999"
outtraj ligands_BEN.pdb onlyframes 1
unstrip
strip ":1,3-999999"
outtraj ligands_FEN.pdb onlyframes 1
_EOF
cpptraj -p complex_vdw_bonded.parm7 <<_EOF
trajin complex_vdw_bonded.rst7
strip ":1,2"
outtraj complex_solvated.pdb onlyframes 1
unstrip
strip ":2-999999"
outtraj complex_BEN.pdb onlyframes 1
unstrip
strip ":1,3-999999"
outtraj complex_FEN.pdb onlyframes 1
_EOF
再次使用tleap生成decharge,vdw和recharge的文件,decharge是配体1,recharge是配体2,修改阅读的pdb的名字即可。
tleap -f- <<_EOF
# load the AMBER force fields
source leaprc.protein.ff19SB
source leaprc.gaff2
source leaprc.water.tip3p
loadAmberParams frcmod.ionsjc_tip3p
loadOff BEN.lib
loadOff FEN.lib
loadamberparams BEN.gaff2.frcmod
loadamberparams FEN.gaff2.frcmod
# coordinates for solvated ligands as created previously by MD
lsolv = loadpdb ligands_solvated.pdb
lbnz = loadpdb ligands_BEN.pdb
lphn = loadpdb ligands_FEN.pdb
# coordinates for complex as created previously by MD
csolv = loadpdb complex_solvated.pdb
cbnz = loadpdb complex_BEN.pdb
cphn = loadpdb complex_FEN.pdb
# decharge transformation
decharge = combine {lbnz lbnz lsolv}
setbox decharge vdw
savepdb decharge ligands_decharge.pdb
saveamberparm decharge ligands_decharge.parm7 ligands_decharge.rst7
decharge = combine {cbnz cbnz csolv}
setbox decharge vdw
savepdb decharge complex_decharge.pdb
saveamberparm decharge complex_decharge.parm7 complex_decharge.rst7
# recharge transformation
recharge = combine {lphn lphn lsolv}
setbox recharge vdw
savepdb recharge ligands_recharge.pdb
saveamberparm recharge ligands_recharge.parm7 ligands_recharge.rst7
recharge = combine {cphn cphn csolv}
setbox recharge vdw
savepdb recharge complex_recharge.pdb
saveamberparm recharge complex_recharge.parm7 complex_recharge.rst7
quit
_EOF
生成好这些过程的文件后,使用setup_run.sh来产生三个步骤的输入文件,在修改setup_run.sh时,注意以下部分。
decharge_crg=":2@H6"
vdw_crg=":1@H6|:2@O1,H6"
recharge_crg=":1@O1,H6"
decharge="ifsc=0,crgmask='$decharge_crg',"
vdw_bonded="ifsc=1,scmask1=':1@H6',scmask2=':2@O1,H6',crgmask='$vdw_crg'"
recharge="ifsc=0,crgmask='$recharge_crg',"
适配修改,注意H6的都改成初始的ambmask,O1,H6的都改成目标的ambmask,:前面的1或者2不要改。
如有必要修改λ,改变prod.tmpl和setup.sh中的值(0.00922开始的那一串)。
该文件将直接生成所需的slurm文件,并提交到对应的机器上,默认使用g-v100-1,运行pmemd.cuda,大致运行时间1小时左右。
有时候pmemd.cuda会运行失败,此时转用cpu来运行,使用run_mpi.py,提交命令:
python run_mpi.py ligands 500000 mpi
将会检查所有ligands下的文件,对于5分钟内没更新,且info中运行步骤在500000 以下的,会提交到32核CPU机器上运行后续的模拟,直到结束。
这个运行步骤非常缓慢,使用32核CPU算完1纳秒的步骤可能需要7-8天,可以尝试先运行一段CPU,再运行一段GPU,改为python run_mpi.py ligands 500000 cuda即可。
运行结束后,使用alchemical-analysis/alchemical_analysis/alchemical_analysis.py来分析结果,注意该文件在python2下运行。
pip2 install matplotlib
pip2 install scipy
pip2 install numpy
pip2 install pymbar==3.0.3
运行:
mkdir -p ana_recharge && cd ana_recharge
../../alchemical-analysis/alchemical_analysis/alchemical_analysis.py -a AMBER -d . -p ../recharge/[01]*/ti00[1-9] -q out -o . -t 300 -v -r 5 -u kcal -f 50 -g -w
mkdir -p ../ana_decharge && cd ../ana_decharge
../../alchemical-analysis/alchemical_analysis/alchemical_analysis.py -a AMBER -d . -p ../decharge/[01]*/ti00[1-9] -q out -o . -t 300 -v -r 5 -u kcal -f 50 -g -w
mkdir -p ../ana_vdw && cd ../ana_vdw
../../alchemical-analysis/alchemical_analysis/alchemical_analysis.py -a AMBER -d . -p ../vdw_bonded/[01]*/ti00[1-9] -q out -o . -t 300 -v -r 5 -u kcal -f 50 -g -w
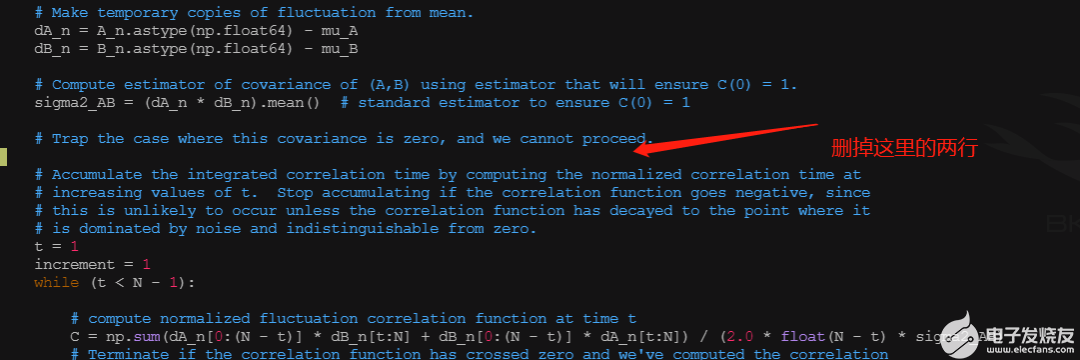
此时只能输出三种变化的结果,将其TI 一列加和得到最终的结果。
如果见到pymbar的warning,只要注释掉对应的assert就可以了。
vim /home/cloudam/.local/lib/python2.7/site-packages/pymbar/timeseries.py
去第162行。

该应用场景解决将蛋白口袋内的残基A变为残基B所产生的相对自由能变。
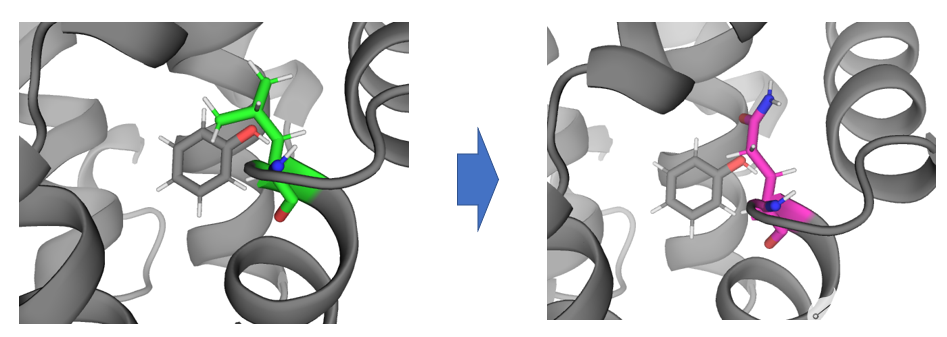
将蛋白口袋内的LEU转化GLN。
首先,使用pymol将分子打开,并选中残基,使用wizard-mutagenesis-protein完成突变,或者使用命令行完成突变:
load*.pdb
cmd.wizard("mutagenesis")
cmd.do("refresh_wizard")
cmd.get_wizard().set_mode("GLN")
cmd.get_wizard().do_select("86/")
cmd.get_wizard().apply()
cmd.set_wizard("done")
save*out_name.pdb,enabled
将突变后的蛋白文件保存为pdb文件。
将突变前的蛋白(WT.pdb)和突变后的蛋白(L86Q.pdb)的pdb文件上传。使用tleap,读取野生型结构。
tleap
source leaprc.protein.ff14SB
source leaprc.gaff2
source leaprc.water.tip3p
loadamberparams frcmod.ionsjc_tip3p
zn = loadpdb WT.pdb
check zn
solvateBox zn TIP3PBOX 10
addIons znCl- 0
savepdb zn box_check.pdb
quit
记录盒子的范德华半径,并将结构中的水提取出来备用。
python dry_for_TI.py box_check.pdb wat.pdb WT_receptor.pdb
同样的方式读取突变型结构,使其保持Amber的原子顺序。
tleap
source leaprc.protein.ff14SB
source leaprc.gaff2
source leaprc.water.tip3p
loadamberparams frcmod.ionsjc_tip3p
zn = loadpdb L86Q.pdb
check zn
solvateBox zn TIP3PBOX 10
addIons zn Cl - 0
savepdb zn L86Q_leap.pdb
quit
python dry_for_TI.py L86Q_leap.pdb wat1.pdbL86Q_dry.pdb
对比两个去水后的文件,发现由于Amber重编号,突变的残基变为84位,此时使用check_diff_online.py,来保证不是突变的残基的位置都绝对一致,以允许进行TI过程。
python check_diff_online.py L84Q L86Q_dry.pdb WT_receptor.pdb 84 L86Q_check.pdbWT_
print的是空字典,说明所有的原子都是匹配的。
再次用tleap读取受体和配体(之前第一部分保存的mol2文件),并读取水盒子,其中ligand是刚才保存的配体(不变部分),m1和m2分别是突变前后的部分(注意突变只改变侧链,不改变主链),注意这里使用了刚才记录的范德华半径。
tleap
source leaprc.protein.ff14SB
source leaprc.gaff2
loadOff FEN.lib
loadamberparams FEN.gaff2.frcmod
source leaprc.water.tip3p
loadamberparams frcmod.ionsjc_tip3p
ligand = loadmol2 FEN.gaff2.mol2
m1 = loadpdb L86Q_check.pdb
m2 = loadpdb WT_check.pdb
w = loadpdbwat1.pdb
protein = combine {m1 m2 w}
complex = combine {m1 m2 ligand w}
set default nocenter on
setBox protein vdw{39.415 43.577 52.292}
savepdb protein protein.pdb
saveamber parmprotein protein.parm7 protein.rst7
setBox complex vdw {39.415 43.577 52.292}
savepdb complex complex.pdb
saveamberparm complex complex.parm7 complex.rst7
quit
使用parmed处理protein.parm7和complex.parm7,以保证正确的所改变的位置提供给TI运算,此处的162为WT或者突变体的残基数,@后面的内容是python get_mutation.py L86Q得到的mapping结果,是在那个突变残基上但也没有变化的残基,84是突变位置,246是84+162。
parmed protein.parm7 <<_EOF
loadRestrt protein.rst7
setOverwrite True
tiMerge :1-162 :163-324 :84&!@CA,C,O,N,H,HA,CB :246&!@CA,C,O,N,H,HA,CB
outparm merged_L86Q_protein.parm7 merged_L86Q_protein.rst7
quit
_EOF
parmed complex.parm7 <<_EOF
loadRestrt complex.rst7
setOverwrite True
tiMerge :1-162 :163-324 :84&!@CA,C,O,N,H,HA,CB :246&!@CA,C,O,N,H,HA,CB
outparm merged_L86Q_complex.parm7 merged_L86Q_complex.rst7
quit
_EOF
正确获得这些文件后,使用:
python auto_gene_inp_run.py 84 162 CA,C,O,N,H,HA,CB L86Q
来生成tmpl文件(run.tmpl文件需要上传),并将tmpl文件转成真正的ti文件放进文件夹,同时使用slurm提交最小化,加热和运行步骤。
注意,这里直接使用cuda很容易断,可以适当自己修改之前的run_mpi.py来使用cpu续跑中断的模拟。
运行结束后的分析略。
 本案例将实际运行一个蛋白-蛋白相互作用上的突变。我们计算新冠病毒受体结合区域(rbd)到人ACE2受体(ace2)复合物上发生Omicron的突变之一的返回突变A484E后的结合自由能变化。
本案例将实际运行一个蛋白-蛋白相互作用上的突变。我们计算新冠病毒受体结合区域(rbd)到人ACE2受体(ace2)复合物上发生Omicron的突变之一的返回突变A484E后的结合自由能变化。
生成连接了二硫键的大复合体水盒,记录vdw盒子大小,额外加入0.15M/L NaCL (总水数量*0.002772)。
tleap
source leaprc.protein.ff14SB
source leaprc.gaff
source leaprc.water.tip3p
loadamberparams frcmod.ionsjc_tip3p
zn = loadpdb omi_SS.pdb
bond zn.333.SG zn.358.SG
bond zn.376.SG zn.429.SG
bond zn.388.SG zn.522.SG
bond zn.477.SG zn.485.SG
bond zn.637.SG zn.645.SG
bond zn.848.SG zn.865.SG
bond zn.1034.SG zn.1046.SG
checkzn
solvateBox zn TIP3PBOX 10
addIons zn Na+ 0
addIons znNa+8 0
addIons znCl- 0
savepdb zn box_check.pdb
quit
python dry_for_TI.py box_check.pdb wat1.pdb omi_rbd.pdb,omi_ace2.pdb
同时生成一个小的水盒,用于跑蛋白部分的TI。
tleap
source leaprc.protein.ff14SB
source leaprc.gaff
source leaprc.water.tip3p
loadamberparams frcmod.ionsjc_tip3p
m1 = loadpdb omi_rbd.pdb
bond m1.4.SG m1.29.SG
bond m1.47.SG m1.100.SG
bond m1.59.SG m1.193.SG
bond m1.148.SG m1.156.SG
check m1
solvateBox m1 TIP3PBOX 10
addIons m1 Na+ 0
addIons m1Na+ 28
addIons m1Cl- 0
savepdb m1 ligands_recharge.pdb
quit
python dry_for_TI.py ligands_recharge.pdb rbd_wat.pdb test_rbd.pdb
检查输入的是否在TI区域之外没有区别,注意输入的顺序,前面是突变后的蛋白,后面是原始的蛋白。
python check_diff_online.py A152EA484E_rbd.pdb omi_rbd.pdb 152 A484E_check.pdb omi_check.pdb
设定正确的二硫键,分别加载两种水盒,输出protein和complex的拓扑学文件和坐标文件。
tleap
source leaprc.protein.ff14SB
source leaprc.gaff
source leaprc.water.tip3p
loadamberparams frcmod.ionsjc_tip3p
ligand = loadpdbomi_ace2.pdb
bond ligand.308.SG ligand.316.SG
bond ligand.519.SG ligand.536.SG
bond ligand.705.SG ligand.717.SG
m1 = loadpdb omi_check.pdb
bond m1.4.SG m1.29.SG
bond m1.47.SG m1.100.SG
bond m1.59.SG m1.193.SG
bond m1.148.SG m1.156.SG
m2 = loadpdb A484E_check.pdb
bond m2.4.SG m2.29.SG
bond m2.47.SG m2.100.SG
bond m2.59.SG m2.193.SG
bond m2.148.SG m2.156.SG
w1 = loadpdb rbd_wat.pdb
w2 = loadpdb wat1.pdb
protein = combine {m1m2w1}
complex = combine {m1m2ligandw2}
set default nocenter on
setBox protein vdw{43.21553.42159.922}
savepdb protein protein.pdb
saveamber parmprotein protein.parm7 protein.rst7
setBox complex vdw{64.171 75.490 114.587}
savepdb complex complex.pdb
saveamberparm complex complex.parm7 complex.rst7
quit
使用parmed处理protein.parm7和complex.parm7,以保证正确的位置。
parmedprotein.parm7 <<_EOF
loadRestrt protein.rst7
setOverwrite True
tiMerge :1-193 :194-386 :152&!@CA,C,O,N,H,HA,CB :345&!@CA,C,O,N,H,HA,CB
outparm merged_A484E_protein.parm7 merged_A484E_protein.rst7
quit
_EOF
parmed complex.parm7 <<_EOF
loadRestrt complex.rst7
setOverwrite True
tiMerge :1-193 :194-386 :152&!@CA,C,O,N,H,HA,CB :345&!@CA,C,O,N,H,HA,CB
outparm merged_A484E_complex.parm7 merged_A484E_complex.rst7
quit
_EOF
正确获得这些文件后,使用:
python auto_gene_inp_run.py 152 193 CA,C,O,N,H,HA,CB A484E
来生成tmpl文件(run.tmpl文件需要上传),并将tmpl文件转成真正的ti文件放进文件夹,同时使用slurm提交最小化,加热和运行步骤(5ns)。
